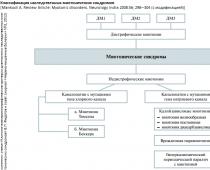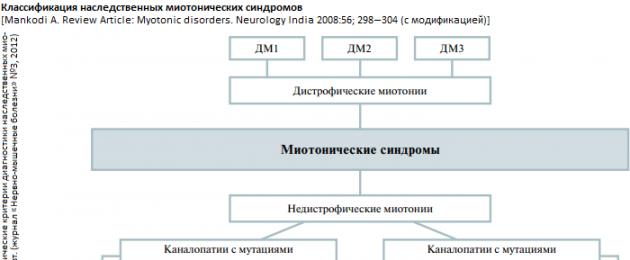
Наследственные миотонические синдромы (НМС) - группа генетически гетерогенных заболеваний ионных каналов хлора и натрия (каналопатии), характеризующиеся повышенной возбудимостью мембраны мышечных волокон, проявляющиеся миотоническими феноменами с постоянной или транзиторной слабостью скелетной мускулатуры. В современной классификации НМС представлены различными генетическими формами [1 ] дистрофических (ДМ) и [2 ] недистрофических миотоний (НДМ).


о клинической и ЭМГ феноменологии миотонии
вы можете прочитать в статье «Клинико-диагностические критерии миотонии» Н. А. Шнайдер; ГБОУ ВПО Красноярский государственный медицинский университет имени проф. В. Ф. Войно-Ясенецкого Министерства здравоохранения РФ, кафедра медицинской генетики и клинической нейрофизиологии ИПО (журнал «Сибирское медицинское обозрение» №3, 2016) [читать ]
Недистрофические миотонии . Самая распространенная форма НДМ - врожденная миотония (ВМ), с распространенностью от 1 до 9,4 на 100 тыс. населения, что зависит от страны и этнической принадлежности. Происхождение ВМ обусловлено мутациями в гене хлорного канала CLCN1 (локус 7q35) в поперечно-полосатых мышцах (нарушается проводимость ионов Cl- в клетку, что ведет к повышенной возбудимости мышечной мембраны и мышечной ригидности). ВМ клинически проявляются:
[1
] генерализованными миотоническими феноменами;
[2
] гипертрофией скелетной мускулатуры;
[3
] транзиторной слабостью;
[4
] дебютом в раннем возрасте;
[5
] стационарным течением и благоприятным прогнозом.
На сегодня выделяют 2 формы ВМ. Первой описана миотония Томсена (МТ) с аутосомно-доминантным типом наследования и лишь 100 лет спустя - миотония Беккера (МБ) с аутосомно-рецессивным типом наследования. Молекулярно-генетические данные показали, что ранее считавшаяся редкой ВМ Беккера встречается даже чаще, чем ВМ Томсена. При дебюте в раннем возрасте пациенты с МТ и МБ имеют, как было указано выше, гипертрофию скелетных мышц («атлетическое сложение», более характерное для ВМ Томсена), иногда легкую миалгию, слабость, задержку мышечного расслабления, а также относительно стационарное течение и благоприятный прогноз. То есть больные с МТ и МБ имеют однотипные клинические проявления, различаясь лишь степенью их выраженности (ВМ Беккера считают несколько более тяжелой, но не в каждом случае).
Дистрофические миотонии
(или ДМ, или сongenital myotonic dystrophy, myotonic dystrophy - DM) является мультисистемным заболеванием, при котором мутация затрагивает развитие и функционирование различных органов и тканей: гладкой и скелетной мышечной ткани, сердца, органа зрения (глаза), головного мозга. Это наиболее распространенное заболевание из класса миотоний. Клиническая картина дистрофической миотонии складывается из 3 синдромов:
[1
] миотонический синдром;
[2
] дистрофический синдром;
[3
] синдром вегетативно-трофических нарушений.
Таким образом, ключевая особенность ДМ - сочетание миотонии, которая характеризуется отсроченным расслаблением после мышечного сокращения, и прогрессирующей мышечной слабости, дистрофии (атрофии). МД характеризуется варьиру ющим началом заболевания: от пренатального периода до 50 - 60 лет. Различают четыре формы по возрастному «пику» начала заболевания: [1 ] врожденная (син.: конгенитальная ДМ или congenital myotonic dystrophy ; клиническая симптоматика развивается сразу после рождения), [2 ] юношеская (син.: ювенильная МД или juvenile DM; с дебютом от 1 года до подросткового возраста), [3 ] классическая (син.: ДМ взрослых или adult DM; - с дебютом у индивидуумов старше 20, но моложе 40 лет) и [4 ] минимальная (син.: МД с поздним дебютом или DM with late onset; у индивидуумов старше 40 лет [как правило, до 60 лет] и с более легким течением). Это объясняется различиями в числе тринуклеотидных повто ров (CTG) в локусе гена МД, кодирующего синтез миотонин-протеинкиназы. Кроме различия в возрасте начала проявления болезни имеются достаточно различные клинические признаки разных подтипов этого заболевания.
До 1994 года ДМ считалась однородным заболеванием. Однако в последние годы после идентификации различных мутаций при сходной клинической симптоматике, напоминающей дистрофическую миотонию, было показано, что это гетерогенное заболевание, представленное тремя подтипами: DM1 (мутация 19q13.3), DM2 (мутация 3q21) и DM3 (мутация 15q21-q24). Существуют отдельные исследования, подтверждающие наличие четвертого подтипа DM и др. (DM4, DMX). Наиболее часто встречается (среди всех миотоний, как среди ДМ, так и среди НДМ) DM1 - ДМ Россолимо-Куршмана-Штейнерта-Баттена (с частотой 13,5 на 100000 живых новорожденных; распространенность в больших популяциях около 1: 8000). Распространенность DM2 (болезнь Thornton-Griggs-Moxley) и DM3 в настоящее время недостаточно изучена.

читайте также пост: Креатинкиназа (справочник невролога)
(на сайт)
В отличие от большинства других наследственных нервно-мышечных заболеваний (и от других форм миотоний, в частности) клинические проявления ДМ вариабельны и могут различаться от человека к человеку даже в пределах одной семьи. Больные с ДМ могут сталкиваться с большим числом разнообразных (необычных для миотонии) проблем, к которым можно отнести низкий уровень жизненной активности, пассивность, депрессию, облысение, нарушения со стороны желудочно-кишечного тракта, сексуальные проблемы, что в ряде случаев приводит к возникновению недопонимания и даже к конфликтным ситуациям между пациентом и лечащим врачом. Следует помнить, что пассивное поведение пациента является по большей мере реальным проявлением данного заболевания, а не желанием человека.
подробнее о DM1, DM2 и DM3 читайте [1 ] в статье «Клинико-генетическая гетерогенность дистрофической миотонии» Н.А. Шнайдер, Е.А. Козулина, Д.В. Дмитренко; Кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования ГОУ ВПО «Красноярская гос. мед. академия Федерального агентства по здраво-охранению и социальному развитию», Россия (Международный неврологический журнал, №3, 2007) [читать ] (или [читать ]) и [2 ] в статье «Миотоническая дистрофия. Современное представление и собственное наблюдение» Т.И. Стеценко, Национальная медицинская академия последипломного образования имени П.Л. Шупика, г. Киев, Украина (журнал «Современ-ная педиатрия» №1, 2014) [читать ]
Обратите внимание ! Для НДМ не характерно развитие парезов конечностей и мышечных атрофий в отличие от больных с ДМ1, у которых вялые дистальные парезы рук и ног служат главной причиной инвалидизации. Тем не менее, у больных с НДМ (форма Томсена, Беккера) специфическими симптомами могут выступать исходная неловкость и мышечная слабость в кистях, обусловленная типичными миотоническими задержками и исчезающая после нескольких повторных произвольных сокращений мышц. Симптом получил наименование «транзиторная слабость», а уменьшение выраженности миотонии при повторных мышечных сокращениях - «феномен врабатывания». Транзиторная слабость отсутствует у больных с ДМ1, а слабость в мышцах кисти остается неизменной даже после уменьшения миотонических проявлений. Наличие транзиторной слабости у больных с НДМ связывали с дебютом ДМ1, что затрудняло дифференциальную диагностику, приводило к неверным нозологическим трактовкам и неадекватным генетическим прогнозам в семьях пробандов. Все это послужило поводом к необходимости выработки объективных критериев наличия или отсутствия транзиторной слабости. Проведение теста с ритмической стимуляцией (РС) с частотой 10 - 60 Гц у больных с НМС выявило декремент М-ответа с восстановлением его амплитуды после тетанизации. Кроме того, при проведении РС у больных с НМС на примере мышц кисти, иннервируемых локтевым нервом, выявляется зависимость между величиной декремента амплитуды М-ответов и выраженностью транзиторной слабости. Выявленный декремент М-ответа при РС в генотипированной группе больных с НДМ объясняется нарушением функции хлорных каналов. [!!! ] Таким образом, декремент амплитуды М-ответов может быть информативным показателем в алгоритме диагностического поиска мутаций в гене хлорного канала у пациентов с НМС.

Читайте также :
статья «Миотоническая дистрофия: генетика и полиморфизм клинических проявлений» Е.О. Иванова, А.Н. Москаленко, Е.Ю. Федотова, С.А. Курбатов, С.Н. Иллариошкин; ФГБНУ «Научный центр неврологии», Москва, Россия; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр», Воронеж (журнал «Анналы клинической и экспериментальной неврологии» №1, 2019) [читать ];
статья «Клинико-электромиографические критерии диагностики наследственных миотонических синдромов» В.П. Федотов, С.А. Курбатов, Е.А. Иванова, Н.М. Галеева, А.В. Поляков; Воронежская медико-генетическая консультация, БУЗ ВО «Воронежская областная клиническая больница №1»; ФГБУ «Медико-генетический научный центр» РАМН, Москва (журнал «Нервно-мышеч-ные болезни» №3, 2012) [читать ];
статья «Случай миотонии Беккера с псевдодоминантным типом наследования: современные подходы к дифференциальной диагностике миотоний Томсена и Беккера» С.А. Курбатов, С.C. Никитин, С.Н. Иллариошкин, П. Гундорова, А.В. Поляков; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр»; Региональная общественная организация Общество специалистов по нервно-мышечным болезням», Медицинский центр «Практическая неврология», Москва; ФГБНУ «Научный центр неврологии», Москва; ФГБНУ «Медико-генетический научный центр», Москва (журнал «Нервно-мышечные болезни» №1, 2016) [читать ];
статья «Случай миотонии Беккера с эквинной деформацией стоп» Г.Е. Руденская, Е.А. Иванова, Н.М. Галеева; Медико-генети-ческий научный центр РАМН, Москва (Журнал неврологии и психиатрии, №8, 2012) [читать ];
статья «Миотонии» (сокращенное изложение) Teruyuki Kurihara, Internal Medicine 2005; 44:1027-1032 (подготовил Ю. Матвиенко) [читать ];
диссертация на соискание ученой степени кандидата биологических наук «Генетическая вариабельность локуса миотонин-протеинкиназы в якутской популяции» Степанова С.К., ФГБНУ «Якутский научный центр комплексных медицинских проблем» ФГБНУ «Научно-исследовательский институт медицинской генетики»; Томск, 2015 [читать ];
статья «Миотоническая дистрофия 2-го типа» Г.Е. Руденская, А.В. Поляков; ФГБУ «Медико-генетический научный центр» РАМН, Москва (журнал «Анналы клинической и экспериментальной неврологии» №2, 2012) [читать ];
статья «Дистрофическая миотония Россолимо-Штейнерта-Куршмана» Авдей Г.М., Кулеш С.Д., Шумскас М.С., Авдей С.А.; УО «Гродненский государственный медицинский университет»; УЗ «Гродненская областная клиническая больница», г. Гродно (Уральский медицинский журнал, №9, 2014) [читать ];
статья «Случай дистрофической миотонии 1-го типа с утяжелением клиники по линии отца» Курбатов С.А., Федотов В.П., Галеева Н.М., Забненкова В.В., Поляков А.В.; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр»; БУЗ ВО «Воронежская областная клиническая больница № 1»; ФГБНУ «Медико-генетический научный центр», Москва (журнал «Анналы клинической и экспериментальной неврологии» №2, 2015) [читать ];
статья «Случай поздней диагностики дистрофической миотонии Россолимо-Штейнерта-Куршмана» Ю.Н. Быков, Ю.Н. Васильев, О.П. Панасюк, А.Ч. Янгутова; Иркутский государственный медицинский университет, кафедра нервных болезней (Сибирский медицинский журнал, №2, 2016) [читать ];
статья «Клинико-генетическая гетерогенность хондродистрофической миотонии» Н.А. Шнайдер, Красноярский ГМУ им. проф. В.Ф. Войно-Ясенецкого, Красноярск (журнал «Нервно-мышечные болезни» №2, 2012) [читать ];
статья «Профилактика аспирационной пневмонии у больных дистрофической миотонией с орофарингеальной дисфагией» Е.А. Бахтина, Н.А. Шнайдер, Т.Л. Камоза, Е.А. Козулина; Красноярская государственная медицинская академия им. В.Ф. Войно-Ясенецкого, кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования; Красноярский государственный торгово-экономический институт, кафедра технологии питания (журнал «Сибирское медицинское обозрение» №3, 2008) [читать ];
статья «Мультидисциплинарный подход к ведению беременных, больных дистрофической миотонией» Н.А. Шнайдер, Е.А. Козулина, В.А. Шульман, С.Ю. Никулина, Р.А. Бутьянов, Е.А. Бахтина; Красноярская государственная медицинская академия им. В.Ф. Войно-Ясенецкого Росздрава; кафедра медицинской генетики и клинической нейрофизиологии ИПО; кафедра внутренних болезней №1 (журнал «Сибирское медицинское обозрение» № 4, 2008) [читать ]
© Laesus De Liro
Любой родитель желает только одного – чтобы малыш родился здоровым и не имел никаких серьезных отклонений в развитии или заболеваний. К огромному сожалению, даже сильного желания не достаточно для того, чтобы избежать возможных проблем – каждая болезнь имеет свою статистику, и порой цифры крайне неутешительны. Узнать о том, что у малыша проблема со здоровьем еще во время его внутриутробного развития порой просто невозможно, многие патологии обнаруживают себя уже после появления на свет. Не исключение и миотонический синдром, входящий в группу заболеваний нервно-мышечного типа.
Некоторые специалисты не классифицируют данный синдром как болезнь, а говорят о нем как о симптоматическом проявлении мышечных нарушений. У пациентов с таким диагнозом нарушена способность мышц к быстрой релаксации после очередного напряжения, то есть процесс расслабления проходит намного дольше, чем у здорового человека. При наличии такой патологии движения ребенка, особенно первые, даются огромными усилиями. Как распознать наличие болезни у малыша и как помочь ему? Это и попробуем выяснить.
Разновидности и причины появления синдрома
При миотоническом синдроме мышцы не способны быстро восстанавливаться
На сегодняшний день группу миотонических заболеваний описывают с разных точек зрения. Так, некоторые включают в нее исключительно врожденные формы миотонии, а некоторые – любые нарушения, касающиеся способности мышц к расслаблению, даже те, что носят временный характер. Классическая форма миотонического синдрома – врожденная. Точные механизмов ее появления у детей не установлено, но выделены предполагаемые причины ее возникновения:
- наследственный фактор, то есть особенности мышечного тонуса передаются ребенку от родителей или кровных родственников;
- нарушения обмена веществ у будущей мамы;
- прием определенных медикаментозных препаратов во время беременности.
Приобретенная форма может становиться следствием малоактивного образа жизни ребенка, перенесенного в раннем возрасте рахита, нарушений нормального обмена веществ в организме. Комплекс мышечных нарушений в рамках миотонического синдрома свойственен в разных своих формах следующим заболеваниям:
- синдром ригидного человека;
- синдром Шварца-Джампела;
- столбняк;
- злокачественная форма гипертермии;
- нейролептический синдром;
- энцефаломиелит.
Характерные для миотонии симптомы появляются также у тех людей, которых укусил ядовитый паук Черная вдова.
Классическая форма миотоничесокго синдрома – это миотония Томпсона. Заболевание носит наследственный характер и передается по аутосомно-доминантному признаку. Начаться оно может в любом возрасте, но зачастую это происходит либо в детстве, либо в период юношества. При наличии патологии в такой форме, наследование в большинстве случаев случается в каждом поколении без исключений, а для передачи болезни детям достаточно наличия проблемы хотя бы у одного родителя.
Миотонический синдром передается по наследству
Вторая форма заболевания, которая встречается реже, – миотония Беккера, передающаяся по аутосомно-рецессивному типу. В такой ситуации болезнь обычно случается лишь в одном поколении на фоне отсутствия подобных проблем в клинической картине семьи . Если оба родителя выступают носителями измененного гена, то с вероятностью в 25% ребенок родится с таким диагнозом. Если же только один из родителей выступает носителем, то есть вероятность того, что чадо также будет носителем, но симптоматических проявлений это давать не будет.
Симптомы проблемы
В основе постановки диагноза лежит специфическое проявление болезни, а именно расстройство двигательного акта. После того, как мышца сократилась, не происходит естественного расслабления, напротив, возникает тоническое напряжение. Сложнее всего даются первые шаги после какого-то периода спокойствия, уже через несколько циклов сокращения-расслабления процесс проходит немного легче. Фаза расслабления мышцы обычно затягивается до 30-ти секунд.
Впервые наличие врожденной проблемы родители могут заметить не сразу. Ярче всего симптомы проявляются во время активности – подъем по лестнице, быстрая ходьба или прочие движения, которые можно отнести к числу сложно скоординированных. Дети с врожденной миотонией начинают позже ходить и говорить, плохо держат спинку.
Признаки на фото
Даже на слабые внешние раздражители мышцы ребенка с таким диагнозом реагируют ярко, возбудимо. Так, также легкий удар игрушкой или пальцами по определенной группе мышц может привести к сильному спазму – на месте удара возникает напряженный мышечный валик, который очень долго расслабляется.
Заболевание специфическое и, как ни странно, сопровождается мышечной слабостью. Больные дети обычно имеют слабый мышечный корсет спины, что приводит к формированию неправильной осанки, слабые мышцы брюшного пресса, что провоцирует проблемы желудочно-кишечного тракта. Слишком слабые мышцы в зоне лица становятся причинами многих проблем, в том числе нарушений зрения (близорукости) и плохого развития речевой функции.
Проще всего заметить так называемую миотоническую атаку. Например, ребенок пытается встать, но почему-то замирает на какое-то время или вовсе падает, но после нескольких повторных усилий ему все удается подняться, зачастую с помощью какой-либо опоры. Резкие движения сопровождаются более сильными спазмами – когда малыш хочет быстро перейти на бег, то он как бы обездвиживается и часто просто падает словно парализованный. В большинстве случаев, когда ребенок немного «расходится», движения даются ему нормально.
Когда болезнь присутствует уже во взрослом возрасте, то можно отметить ее наличие визуально – подросток выглядит накачанным в тех группах мышц, которые подвержены постоянному напряжению. Это могут быть как мышцы спины или ног, что выглядит эстетично, так и мышцы лица и шеи, что уже не имеет столь привлекательного вида.
Как вылечить миотонический синдром у ребенка
Современная медицина не может на данный момент предложить способа, который бы помог полностью избавиться от проблемы. Но опускать руки не стоит – благодаря симптоматическому лечению ребенок сможет вести нормальный образ жизни. При наличии интенсивных атак назначается курс медикаментозной терапии, который помогает минимизировать их. Если же симптомы не имеют ярких проявлений, то лечение ограничивается следующими мероприятиями:

Для улучшения эффекта врачом могут быть назначены физиотерапевтические процедуры (например, иглорефлексотерапия или электрофорез) и рекомендованы занятия по плаванию.
Диагноз «детский миотонический синдром» - это не приговор, комплексный подход и следование рекомендациям специалистов позволят ребенку нормально развиваться и вести активную жизнь в самостоятельном будущем .
Видео о детском массаже
Здоровые, улыбающиеся дети - самая большая радость для родителей. Но не всегда бывает так, что у заботливых и внимательных пап и мам растут не болеющие малыши.
В педиатрии есть такие состояния, о которых родители просто не знают и не догадываются. Поэтому, когда врач поставит диагноз ребенку «миотонический синдром», то это зачастую является ударом для его родственников. Что же это за состояние, каковы его симптомы, угрозы, и чем лечить болезнь?
Симптомы миотонического синдрома у детей
После того, как родители услышат подобный диагноз, паниковать не нужно, да и времени на это нет. Следует оперативно приниматься за лечение и сделать все, чтобы вылечить ребенка, устранить возникающие угрозы.
Миотонический синдром - заболевание нервно-мышечного характера. Оно характеризуется снижением общего тонуса мышц. Когда они отвечают сокращением на внешние раздражители, то затем с большим трудом расслабляются. Такая мышечная слабость всегда формирует у мальчика или девочки вялую осанку. Дети значительно позже своих сверстников начинают ходить, им тяжело держать спину ровной. Нередко у таких деток наблюдаются нарушения работы суставов. Следует отметить, что пониженный тонус характерен всем мышцам, в том числе и мышцам лица, языка, по этой причине дети и говорить начинают гораздо позже. А если речь развивается вовремя, то с большими нарушениями, невыразительностью.
Слабый мышечный каркас таких детей в зоне пресса приводит также к нарушениям работы желчно-выводящих путей, к запорам. Иногда низкий тонус мышц провоцирует энурез, а слабость глазных мышц - близорукость. Дети с миотоническим синдромом устают быстрее других малышей, они утомляются даже от небольших умственных и физических нагрузок. Это значит, что учиться им будет трудно. Ведь практически регулярно таких деток мучают головные боли. С детства у них развивается сколиоз и остеохондроз, может деформироваться позвоночник, что, конечно же, влияет и на психическое состояние детей.
Читайте также
Все эти признаки миотонического синдрома заметны не сразу после рождения, и, возможно, родителям самим даже трудно вначале диагностировать какие-либо отклонения. Симптомы миотонического синдрома выявляет педиатр. Поэтому следует регулярно посещать с ребенком детскую консультацию, ходить на плановые медицинские осмотры, чтобы вовремя диагностировать возможные проблемы развития малыша.
Причины возникновения
Врачи не зря говорят, что беременность надо планировать. В организме здоровой мамы, которая серьезно относиться к появлению на свет своего первенца, формируются здоровые половые клетки. Прием фолиевой кислоты еще до наступления беременности обезопасит родителей от появления на свет нездорового ребенка, предотвратит пороки его внутриутробного развития.
Причиной митонического синдрома у детей бывают и генетические факторы, а именно особенности тонуса мышц, которые передаются по наследству. Нередко возникновение вышеуказанной болезни у детей - это последствия рахита и нервно-мышечных заболеваний, нарушения обмена веществ и гиподинамия самой мамы в период вынашивания ребенка.
Лечение миотонического синдрома у детей
Прежде всего, нужно организовать здоровый и активный образ жизни малыша после его появления на свет. Это означает кормление грудью и прогулки на свежем воздухе в любое время года, массажи и ежедневную гимнастику, зарядку по утрам. Такие процедуры станут хорошими профилактическими мероприятиями, чтобы избежать дальнейшего прогресса слабости мышц. Но не всегда этих мер бывает достаточно. Речь идет о случаях, когда причиной миотонического синдрома являются генетические факторы. Тогда назначается квалифицированное лечение, к которому привлекаются логопеды и ортопеды, терапевты и окулисты. Для успешного устранения миотонического синдрома у малыша надо прибегнуть к курсу массажа и обязательно пройти полный курс физиотерапевтических процедур. Иногда назначается и медикаментозный курс лечения. Доктора приписывают препараты, улучшающие обменные процессы и повышающие нервно-психическую проводимость.
Можно также лечить детей с миотоническим синдромом в специальных медицинских центрах, где разработаны программы коррекции здоровья. Они включают физиотерапию и занятия с логопедом, психологом, иглорефлексотерапию.
Наследственно-семейное поражение поперечно-полосатой мускулатуры, проявляющееся пролонгированным расслаблением мышц после их сокращения. Кроме типичного миотонического феномена, заболевание характеризуется гипертрофическими изменениями пораженных мышц, манифестацией с поражения кистей, частой вовлеченностью лицевой мускулатуры. Миотония Томсена диагностируется на основании анамнестических данных, результатов неврологического осмотра, ЭМГ и ЭНГ, молекулярно-генетического исследования. Лечение сводится к назначению препаратов, уменьшающих тонические реакции мышц и нормализующих ионное состояние миофибрилл.

Общие сведения
Миотония Томсена входит в группу наследственных миотоний , включающую также миотонию Россолимо-Штейнерта-Куршмана , врожденную парамиотонию Эйленбурга, миотонию Беккера и еще ряд заболеваний. Болезнь Томсена приобрела свое название в соответствии с фамилией ученого, подробно описавшего ее симптомы и порядок наследования на примере 4-х поколений своей семьи. За 2 года до Томсена (в 1874 г.) первое описание заболевания дал Лейден. Поэтому в современной неврологии употребляется также название болезнь Лейдена-Томсена.
У больных с данным видом миотонии прослеживается доминантный тип наследования патологического аутосомного гена. Частота встречаемости заболевания составляет по различным оценкам от 3 до 7 человек на 1 млн. населения.
Этиология и патогенез миотонии Томсена
Миотония Томсена относится к наследственным каналопатиям. Как и миотония Беккера, заболевание связано с дефектом 7-й хромосомы, а именно гена CLCN1, детерминирующего синтез белка хлорных ионных каналов миофибрилл скелетной мускулатуры. Следствием нарушения синтеза этого специфического белка является уменьшенное прохождение ионов хлора внутрь миофибриллы и их скопление на поверхности мембраны мышечного волокна (сарколеммы). Возникающая в результате биоэлектрическая нестабильность сарколеммы обуславливает ее чрезмерную возбудимость. При этом периферический неврон функционирует без отклонений, но на обычный нервный импульс мембрана миофибриллы реагирует повышенным возбуждением, которое препятствует нормальному расслаблению мышечного волокна после его сокращения. Причем, чем быстрее происходит сокращение миофибриллы, тем более затрудненно ее расслабление. Однако после серии сокращений реализуются компенсаторные механизмы, ионные каналы начинают усиленно функционировать и ситуация нормализуется.
Морфологические изменения мышечной ткани неспецифичны для миотонии Томсена и являются типичными для большинства миотоний. Отмечается централизация ядер сарколеммы, увеличение площади сечения миофибрилл, свидетельствующее о их гипертрофии. Электронная микроскопия определяет гипертрофию саркоплазматического ретикулума, увеличение размеров митохондрий и изменение их формы, утолщение телофрагмы.
Симптомы миотонии Томсена
В большинстве случаев манифестация болезни приходится на ранний детский возраст. Ведущим в клинике заболевания является миотонический симптомокомплекс, так называемый миотонический феномен. Он характеризуется пролонгированной мышечной релаксацией после совершения быстрого движения. Клинически это проявляется невозможностью быстро произвести следующее движение из-за тонического спазма мышц, участвовавших в предыдущем двигательном акте. Примечательно, что после серии активных движений скорость мышечного расслабления нормализуется и пациент может двигаться практически как здоровый человек. Перерыв в работе мышц приводит к возобновлению проблем с мышечной релаксацией в начале новой фазы двигательной активности.
Патогномоничным для болезни Томсена является ее манифестация с патологических изменений в кистях. Затем миотонические проявления распространяются на мускулатуру ног, мимическую и жевательную группы мышц. В результате тонические спазмы наблюдаются при резком начале ходьбы, зажмуривании глаз, смыкании зубов и т. п. движениях. При попытке быстрой ходьбы пациенты утрачивают равновесие и могут упасть из-за развития общей скованности. Эмоциональные переживания и пребывание в холоде усиливают миотонические реакции.
Своеобразен внешний вид пациентов с миотонией Томсена. Вследствие диффузной мышечной гипертрофии они зачастую имеют телосложение атлетов. Их мышцы отличаются чрезмерно плотной консистенцией, но имеют сниженную силу. Мышечная гипертрофия является отличительным симптомом заболевания, позволяющим дифференцировать его от миотонической дистрофии Россолимо-Штейнерта-Куршмана, сопровождающейся атрофиями мышц.
Диагностика миотонии Томсена
Поскольку миотония Томсена обычно манифестирует в раннем возрасте, родители заболевшего ребенка обращаются прежде всего к педиатру , который и направляет их на консультацию детского невролога . Для выявления миотонического симптомокомплекса невролог просит обследуемого несколько раз быстро сжать и разжать кулак. Замедленное разжимание пальцев в начале тестирования и постепенная нормализация движений при их повторении свидетельствуют в пользу миотонии. О наличие миотонического феномена говорит образование «валика» локального мышечного сокращения в ответ на постукивание неврологическим молоточком по мышце. В неврологическом статусе выявляется снижение мышечной силы и миотонический характер сухожильных рефлексов. Возможна миотоническая реакция зрачков на световое раздражение.
Основополагающими в диагностике миотонии Томсена являются электрофизиологические исследования : электромиография и электронейрография . Первая выявляет специфичные для миотонии повторяющиеся высокочастотные разряды, а вторая дает возможность полностью исключить поражение анимальной нервной системы.
Биопсия мышц позволяет изучить морфологические изменения мышечной ткани. Тем не менее, выявляемая при болезни Томсена гипертрофия миофибрилл и централизация ядер их сарколеммы наблюдается и при других типах миотоний. Для постановки точного диагноза с верификацией вида миотонии необходимо проведение молекулярно-генетического анализа, позволяющего определить дефект гена CLCN1.
Лечение миотонии Томсена
Современной медициной еще не разработана радикальная терапия генных заболеваний. Поэтому основная задача лечения миотонии Томсена сводится к уменьшению ее проявлений и замедлению прогрессирования симптомов. Для снижения тонической спастичности используются такие препараты, как дифенин, фенитоин, карбамазепин. Для поддержания ионного равновесия мембран миофибрилл применяют препараты кальция в виде медикаментозной терапии, электрофореза , гальванизации воротниковой зоны. С этой же целью рекомендованы мероприятия по уменьшению содержания калия в организме: диета с малым содержанием калия, мочегонная терапия. Из мочегонных средств предпочтительно назначение ацетазоламида, поскольку, кроме усиленного выведения калия с мочой, он также увеличивает проницаемость сарколеммы.
Особое значение в комплексном лечении миотонии Томсена имеют массаж и ЛФК . Они должны проводиться с особой осторожностью, чтобы улучшить обменные процессы мышечной ткани и одновременно уменьшить ее наклонность к тоническому спазмированию.
Прогноз и профилактика
Миотония Томсена сохраняется в течение всей жизни. Доброкачественное течение и медленное прогрессирование болезни делает ее прогноз относительно благоприятным. Пациенты, как правило, сохраняют трудоспособность. Однако, их профессия не должна быть связана с необходимостью совершать быстрые движения. Такие люди не могут работать на конвейере, быть водителями, пожарными, полицейскими и т. п.
К мерам первичной профилактики болезни Томсена можно отнести консультирование генетиком семейной пары, планирующей беременность и имеющей случаи заболевания среди родственников. Профилактикой миотонических приступов служит исключение резкой двигательной активности, переохлаждения , физических перегрузок и эмоционального волнения.
Шамик Виктор Борисович
Шамик Виктор Борисович, Профессор кафедры детской хирургии и ортопедии РостГМУ, Доктор медицинских наук, Член диссертационного совета Ростовского медицинского университета по специальности «детская хирургия», Член ученого совета педиатрического факультета, Врач детский травматолог-ортопед высшей квалификационной категории
Запись на прием к специалисту

Винников Сергей Владимирович
Винников Сергей Владимирович , Детский травматолог-ортопед травматолого-ортопедического отделения для детей поликлиники МБУЗ «Городская больница №20» города Ростова-на-Дону
Запись на прием к специалисту

Фоменко Максим Владимирович
Фоменко Максим Владимирович Кандидат медицинских наук. Заведующий отделением травматологии и ортопедии для детей, кандидат медицинских наук, детский травматолог-ортопед высшей категории
Запись на прием к специалисту

Лукаш Юлия Валентиновна
Лукаш Юлия Валентиновна Кандидат медицинских наук, врач травматолог-ортопед, доцент кафедры детской хирургии и ортопедии
Запись на прием к специалисту
 Редактор страницы: Крючкова Оксана Александровна
Редактор страницы: Крючкова Оксана Александровна


Анатомическое строение приводящих мышщ бедра
Подкожная миотомия приводящих мышц бедер у детей наиболее часто производится по поводу спастических двусторонних контрактур (болезнь Литтла) одновременно с обеих сторон и требует последующего наложения большой гипсовой повязки (обычно от пальцев стоп до реберной дуги) в положении максимального разведения кот. Поэтому для обезболивания целесообразен наркоз, особенно в тех случаях, когда, наряду с устранением приводящих контрактур бедер, предполагают устранить и другие сопутствующие деформации.
Техника выполнения этой операции несложна. Ногу отводят до выраженного напряжения приводящих мышц ниже середины пупартовой связки нащупывают пульсирующую бедренную артерию. На расстоянии 2-3 см кнутри от нее я чуть края лонной кости вкалывают тенотом в продольном направлении через слегка оттянутую в сторону кожу достаточно глубоко. Затем поворачивают тенотом на 90° острием в медиальную сторону и пилящими движениями, продолжая отводить ногу, рассекают все медиально лежащие от тенотома тяжи приводящих мышц, что обычно сопровождается характерный треском. Если ранка кожи очень мала, ее не зашивают, в противном случае накладывают один кетгутовый шов.
Осложнения при подкожной ми отомни аддукторов бедер наблюдаются крайне редко и ограничиваются гематомой с отеком мошонки или гематомой вульвы, которая в течение 7-8 дней полностью рассасывается.
Подкожное удлинение ахиллова сухожилия по Банеру может быть легко выполнено под местным обезболиванием.
Техника операции: чтобы получить максимальное напряжение сухожилия стопе придается максимальная тыльная фиксия. Узким тенотомом в сагиттальном направлении прокалывают кожу и ахиллово сухожилие через всю его толщину на 2-3 см выше его прикрепления к пяточному бугру. Поворачивают тенотом на 90° (кнутри при эквивоварусной и кнаружи при эквиновальгусной деформации) и пилящими движениями пересекают им соответствующую половину сухожилия. Вынимают тенотом, прокалывают им кожу и сухожилие на 4-5 см проксимальнее первого прокола, где аналогичным образом пересекают поперечно половину сухожилия в противоположном направлении. После этого легкой редрессацией благодаря параллельному расположению волокон удается произвести отделение обеих половинок сухожилия друг от друга и устранить деформацию вплоть до гиперкоррекции Z-образным удлинением сухожилия. При спастическом эквинусе во избежание образования пяточной стопы следует ограничиться только коррекцией
Во всех случаях ахиллотомии у детей накладывается гипсовая повязка до средней трети бедра на 4-5 недель в корригированном (при спастическом эквинусе) или гиперкорригированном положении.
Закрытое удлинение ахиллова сухожилия асептичнее и требует меньше времени для его выполнения, чем открытое, в этом его преимущество. Но оно имеет и отрицательные стороны. При закрытом удлинении иногда вместо удлинения производится полное поперечное пересечение сухожилия со значительным расхождением его концов, а регенерация сухожилия даже у детей тем совершеннее, чем меньше расстояние между его концами. При закрытом удлинении, кроме того, не пересекается задняя стенка капсулы голеностопного сустава, которая в ряде случаев (например, при застарелой врожденной косолапости у детей старшего возраста, при артрогрипотической косолапости и др.) оказывается рубцово измененной и продолжает удерживать стопу в положении эквинуса даже после полного пересечения ахиллова сухожилия. Получаемая же в таких случаях последующей редрессацией «гиперкоррекция» является не истинной, а кажущейся, так как она происходит только за счет гиперэкстензии в шопаровском (и отчасти в лисфранковом) суставе; образуется, таким образом, неполноценная в функциональном отношении стопа, так называемая, «стопа-качалка». Поэтому при выборе способа ахиллотомии у детей всегда следует учитывать клиническую картину эквинуса.